I want to create a pool of cells with random point mutations in the H. influenzae murE gene, and to select and screen this pool of cells for hypercompetent mutants. I'm going to do this by mutagenizing the DNA with the chemical mutagen ethyl methanesulfonate (EMS) in vitro and then transforming it into cells, rather than mutagenizing cells.
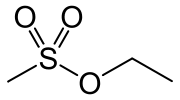
![]() One unanticipated benefit of the in vitro method is that the mutation spectrum is better. With in vivo mutagenesis, EMS produces mainly GG-to-AT transition mutations by alkylating guanines in DNA, creating O-6 ethylguanine which mispairs with T instead of C during DNA replication. (info from Wikipedia). But the in vitro work found a much less biased distribution, with 42% GC-to-AT transitions, 34% AT-to-GC transitions, and 24% GC-toCG transversions.
One unanticipated benefit of the in vitro method is that the mutation spectrum is better. With in vivo mutagenesis, EMS produces mainly GG-to-AT transition mutations by alkylating guanines in DNA, creating O-6 ethylguanine which mispairs with T instead of C during DNA replication. (info from Wikipedia). But the in vitro work found a much less biased distribution, with 42% GC-to-AT transitions, 34% AT-to-GC transitions, and 24% GC-toCG transversions.
Plan:
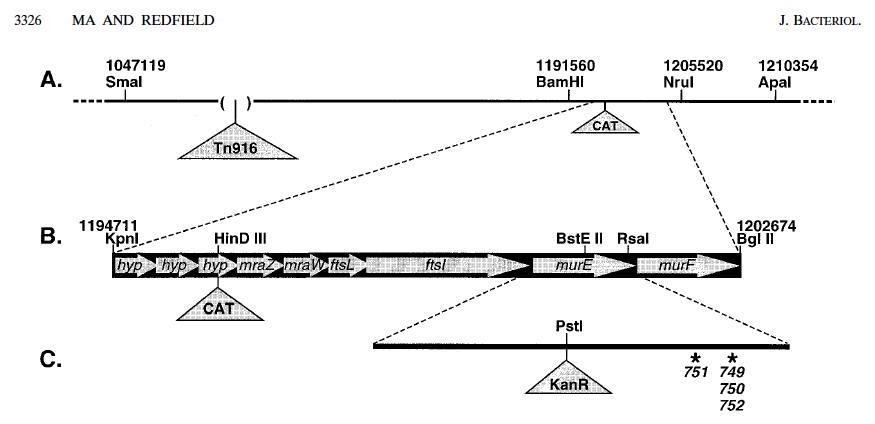
Step 1. Cut chromosomal DNA of strain RR797 with the restriction enzymes KpnI and BglII. This strain has a chloramphenicol resistance cassette inserted about 5 kb away from the known murE mutations, and this digest creates an 8 kb fragment that contains both the CAT cassette and murE.
This step could probably be omitted if necessary, because random fragmentation of the DNA will accomplish almost as much.
Here's the map:
![]()
Step 2. Soak this DNA in an EMS solution for 1 hr.
Step 3. Wash the DNA and transform it into competent wildtype cells. Use about 100 ng DNA per ml, so that each cell is likely to recombine only a single DNA fragment.
Step 4. Select for chloramphenicol resistance, to enrich for cells that have recombined in murE. This will also confirm that the level of DNA damage was not so high as to limit transformation. I should be able to get many thousands of independent transformants.
Step 5. Pool chloramphenicol resistant colonies, creating a separate pool from each plate. I want enough colonies per pool that each is likely to contain at least one hypercompetent mutant - how many colonies will this be? I know of three mutations that produce hypercompetence, which should let me predict the expected frequency. The enrichment can increase the frequency of hypercompetence by 1000-fold, if all the mutants are as hypercompetent as the ones we have. So if the frequency of hypercompetence is 1/1000, I should put about 1000 colonies in each pool. Aim for about 5 pools. Freeze some of the cells of each pool.
Step 6. Grow the pooled cells in sBHI at low density for a dew hours, then transform with cloned or PCR'd NovR DNA (or a different martker?). Plate on nov plates.
Step 7. Screen individual NovR colonies for hypercompetence by touching them to plain plates and then resuspending the rest of the cells in sBHI containing MAP7 DNA and plating on Kan (or Nov?) plates. Do only 10 colonies per pool.
Step 8. For each pool, pick one high-transformation colony from its toothpicked plate, and retest its competence with a simple time course.
First we should test different levels of mutagenesis:
The protocol we have (Lai et al.) says to use 1 µg DNA in 20 µl 10 mM EMS for 1 hr; this gave 5-6 mutations per kb in the clones they sequenced. It also reduced the transformation efficiency of the plasmid insert they mutagenized to about 60%. If they carefully standardized the amounts of DNA, this reduction should have been a direct consequence of DNA damage and repair processes, since they were not selecting for function of their mutagenized insert.
5-6 mutations per kb sounds pretty good for us, since about half of them will be silent, but I think we should first try a wide range of concentrations. For the cell mutagenesis (many years ago) I used 50 mM for 45 min and 80 mM for 30 min (RR expt # 181), but we want much heavier mutagenesis here. So here let's try 0, 2, 5, 10, 20, 50, and 100 mM - that's 7 DNA samples to do transformations with.
Two assays for the extent of mutagenesis:
1. (To identify an optimal concentration) Mutations creating low-level resistance to novobiocin: Mutagenize any novS DNA (e.g. RR797) and transform into KW20 and select for low-level novobiocin resistance (1 µg/ml), to check the efficacy of the mutagenesis. There should be an optimal dose of EMS, above which the frequency of nov resistance drops because the DNA is too damaged to recombine or contains too many mutations that block gene function.
2. (To identify concentrations that are too high) Mutations that inactivate the CAT cassette: Mutagenize RR797 DNA and transform KW20 to chloramphenicol resistance. At some EMS dose the transformation frequency will decrease because the DNA is too damaged to recombine or contains too many mutations that block gene function. (This test could also be done with any point mutation creating antibiotic resistance.)![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
 One unanticipated benefit of the in vitro method is that the mutation spectrum is better. With in vivo mutagenesis, EMS produces mainly GG-to-AT transition mutations by alkylating guanines in DNA, creating O-6 ethylguanine which mispairs with T instead of C during DNA replication. (info from Wikipedia). But the in vitro work found a much less biased distribution, with 42% GC-to-AT transitions, 34% AT-to-GC transitions, and 24% GC-toCG transversions.
One unanticipated benefit of the in vitro method is that the mutation spectrum is better. With in vivo mutagenesis, EMS produces mainly GG-to-AT transition mutations by alkylating guanines in DNA, creating O-6 ethylguanine which mispairs with T instead of C during DNA replication. (info from Wikipedia). But the in vitro work found a much less biased distribution, with 42% GC-to-AT transitions, 34% AT-to-GC transitions, and 24% GC-toCG transversions.Plan:
Step 1. Cut chromosomal DNA of strain RR797 with the restriction enzymes KpnI and BglII. This strain has a chloramphenicol resistance cassette inserted about 5 kb away from the known murE mutations, and this digest creates an 8 kb fragment that contains both the CAT cassette and murE.
This step could probably be omitted if necessary, because random fragmentation of the DNA will accomplish almost as much.
Here's the map:
Step 2. Soak this DNA in an EMS solution for 1 hr.
Step 3. Wash the DNA and transform it into competent wildtype cells. Use about 100 ng DNA per ml, so that each cell is likely to recombine only a single DNA fragment.
Step 4. Select for chloramphenicol resistance, to enrich for cells that have recombined in murE. This will also confirm that the level of DNA damage was not so high as to limit transformation. I should be able to get many thousands of independent transformants.
Step 5. Pool chloramphenicol resistant colonies, creating a separate pool from each plate. I want enough colonies per pool that each is likely to contain at least one hypercompetent mutant - how many colonies will this be? I know of three mutations that produce hypercompetence, which should let me predict the expected frequency. The enrichment can increase the frequency of hypercompetence by 1000-fold, if all the mutants are as hypercompetent as the ones we have. So if the frequency of hypercompetence is 1/1000, I should put about 1000 colonies in each pool. Aim for about 5 pools. Freeze some of the cells of each pool.
Step 6. Grow the pooled cells in sBHI at low density for a dew hours, then transform with cloned or PCR'd NovR DNA (or a different martker?). Plate on nov plates.
Step 7. Screen individual NovR colonies for hypercompetence by touching them to plain plates and then resuspending the rest of the cells in sBHI containing MAP7 DNA and plating on Kan (or Nov?) plates. Do only 10 colonies per pool.
Step 8. For each pool, pick one high-transformation colony from its toothpicked plate, and retest its competence with a simple time course.
First we should test different levels of mutagenesis:
The protocol we have (Lai et al.) says to use 1 µg DNA in 20 µl 10 mM EMS for 1 hr; this gave 5-6 mutations per kb in the clones they sequenced. It also reduced the transformation efficiency of the plasmid insert they mutagenized to about 60%. If they carefully standardized the amounts of DNA, this reduction should have been a direct consequence of DNA damage and repair processes, since they were not selecting for function of their mutagenized insert.
5-6 mutations per kb sounds pretty good for us, since about half of them will be silent, but I think we should first try a wide range of concentrations. For the cell mutagenesis (many years ago) I used 50 mM for 45 min and 80 mM for 30 min (RR expt # 181), but we want much heavier mutagenesis here. So here let's try 0, 2, 5, 10, 20, 50, and 100 mM - that's 7 DNA samples to do transformations with.
Two assays for the extent of mutagenesis:
1. (To identify an optimal concentration) Mutations creating low-level resistance to novobiocin: Mutagenize any novS DNA (e.g. RR797) and transform into KW20 and select for low-level novobiocin resistance (1 µg/ml), to check the efficacy of the mutagenesis. There should be an optimal dose of EMS, above which the frequency of nov resistance drops because the DNA is too damaged to recombine or contains too many mutations that block gene function.
2. (To identify concentrations that are too high) Mutations that inactivate the CAT cassette: Mutagenize RR797 DNA and transform KW20 to chloramphenicol resistance. At some EMS dose the transformation frequency will decrease because the DNA is too damaged to recombine or contains too many mutations that block gene function. (This test could also be done with any point mutation creating antibiotic resistance.)